

Summary Card
Definition of Craniosynostosis
The premature fusion of one or more cranial sutures. It can be isolated or part of a syndrome.
Virchow's Law in Craniosynostosis
Virchow's Law states that premature suture fusion results in cranial growth predominantly parallel to sutures.
Anatomy of Craniosynostosis
5 sutures fuse over a period of time: metopic (6-8 months), sagittal (22 years), coronal (24 years), lambdoidal (26 years), squamosal (60 years).
Classification of Craniosynostosis
By the affected suture(s) or head shape.
Secondary Features of Craniosynostosis
Facial dysmorphology, associated limb abnormalities, and potentially raised intracranial pressure.
Syndromic Craniosynostosis
Majority are autosomal dominant FGFR mutations with abnormal facies and limb abnormalities.
Positional Plagiocephaly
External pressure resulting in a rhomboid-shaped head, flat occiput with ipsilateral prominence of cheeks and brows.
Investigations
X-ray, CT, or MRI can help confirm the clinical diagnosis, identify sutures, assess for complications, and plan for surgery.
Primary Contributor: Dr Abdulrahman Abdulaziz Alghamdi, King Fahad University Hospital.
Definition of Craniosynostosis
Craniosynostosis is the premature fusion of one or more cranial sutures. It can be an isolated abnormality or part of a syndrome. It occurs in ~1 per 2500 births. This is illustrated in the image below.

Risk Factors
There are specific risk factors for developing craniosynostosis. These include:
- Family history
- Advanced maternal age
- Use of clomiphene citrate for infertility
- Pregnancy-related issues: smoking, hydrocephalus, hypothyroidism
- Male gender
Virchow's Law in Craniosynostosis
Virchow's Law states that premature suture fusion results in cranial growth predominantly parallel to sutures.
Virchow's Law states that premature suture fusion results in cranial growth predominantly parallel to sutures. It does not occur perpendicularly.
This means that each fusion pattern has a characteristic skull shape, as illustrated below.
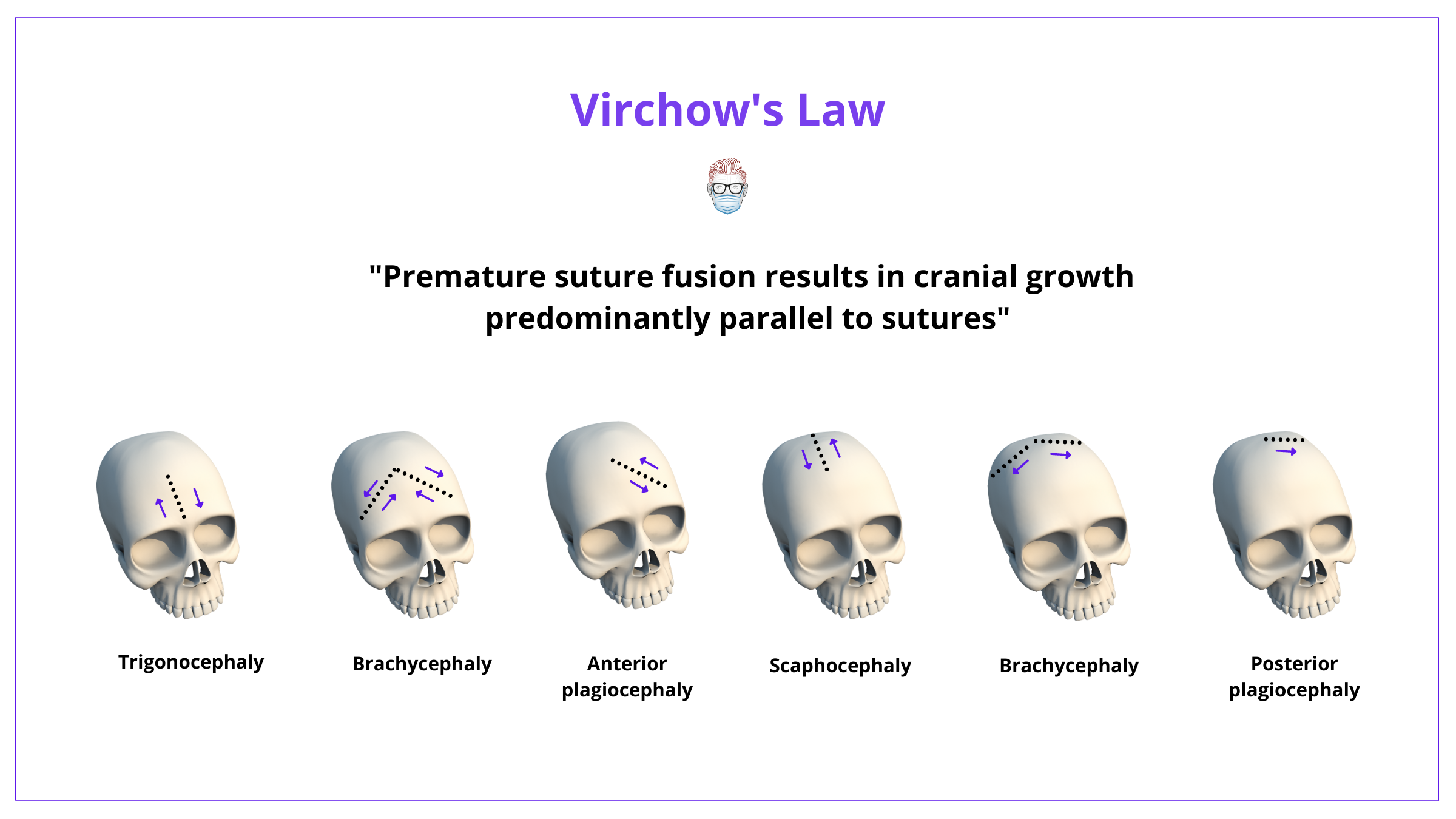
Other theories are published in regard to the pathophysiology and pathoanatomy of craniosynostosis. These include:
- Dural abnormality: suture abnormality relating to the osteoinductive properties of the dura mater (McCarthy).
- Skull base abnormality: abnormalities in the skull base result in abnormal tension on the cranial base exerted through the dura mater (Moss).
Anatomy of Craniosynostosis
Metopic suture is first to fuse at ~6-8 months, followed by sagittal (22 years), coronal (24 years), lambdoidal (26 years) & finally squamosal (60 years).
The skull is constructed of 2 frontal bones, 2 parietal bones, and one occipital bone. In newborns, these bones are separated by patent sutures and fontanelles. The function of these fontanelles and patent sutures:
- Allow for brain growth
- Overlapping of skull bones during labor
The normal cranial suture anatomy can be visualized below.
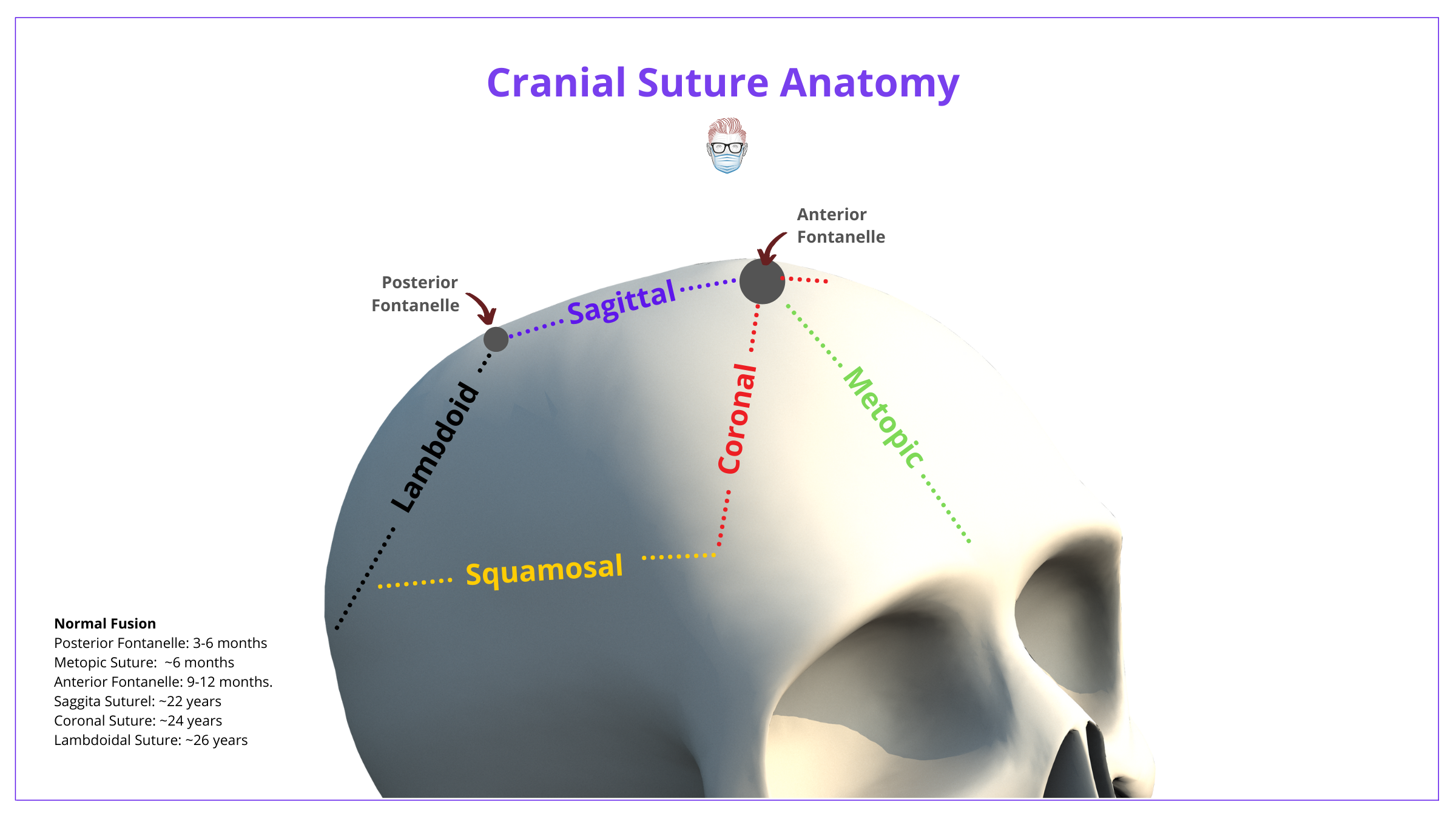
The premature fusion of one or more sutures leads to craniosynostosis. The normal timeline of cranial suture and fontanelle fusion is depicted below.
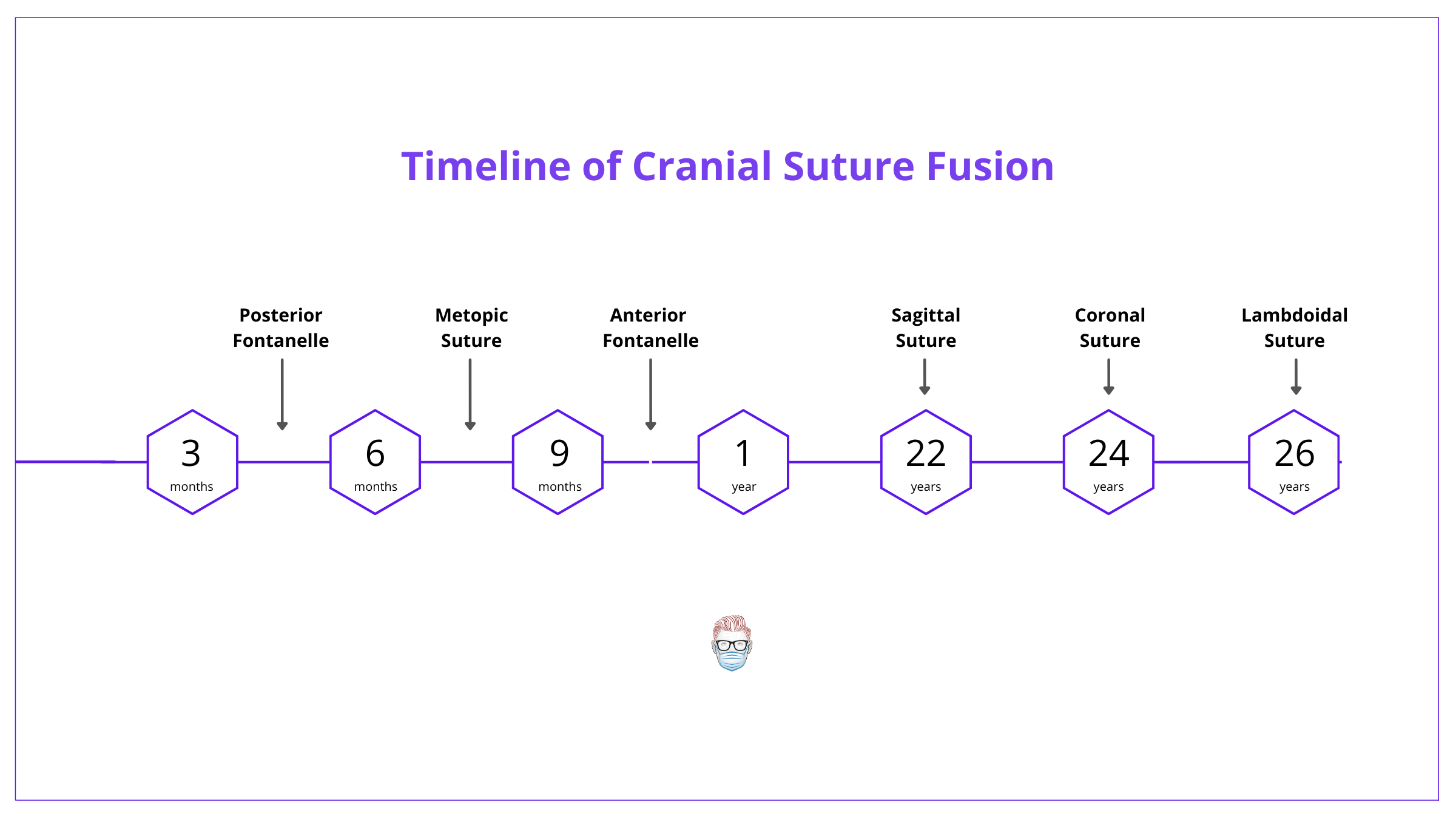
Craniosynostosis usually begins during pregnancy or the first year of life. It's usually completed by 3 years of age. Important dates in "normal" fusions and closures.
- Posterior Fontanelle: 3-6 months
- Metopic: 6-8 months
- Anterior Fontanelle: 9-12 months
- Sagittal: 22 years (most commonly involved)
- Coronal: 24 years
- Lambdoidal: 26 years
- Squamosal: approximately 60 years
Classification of Craniosynostosis
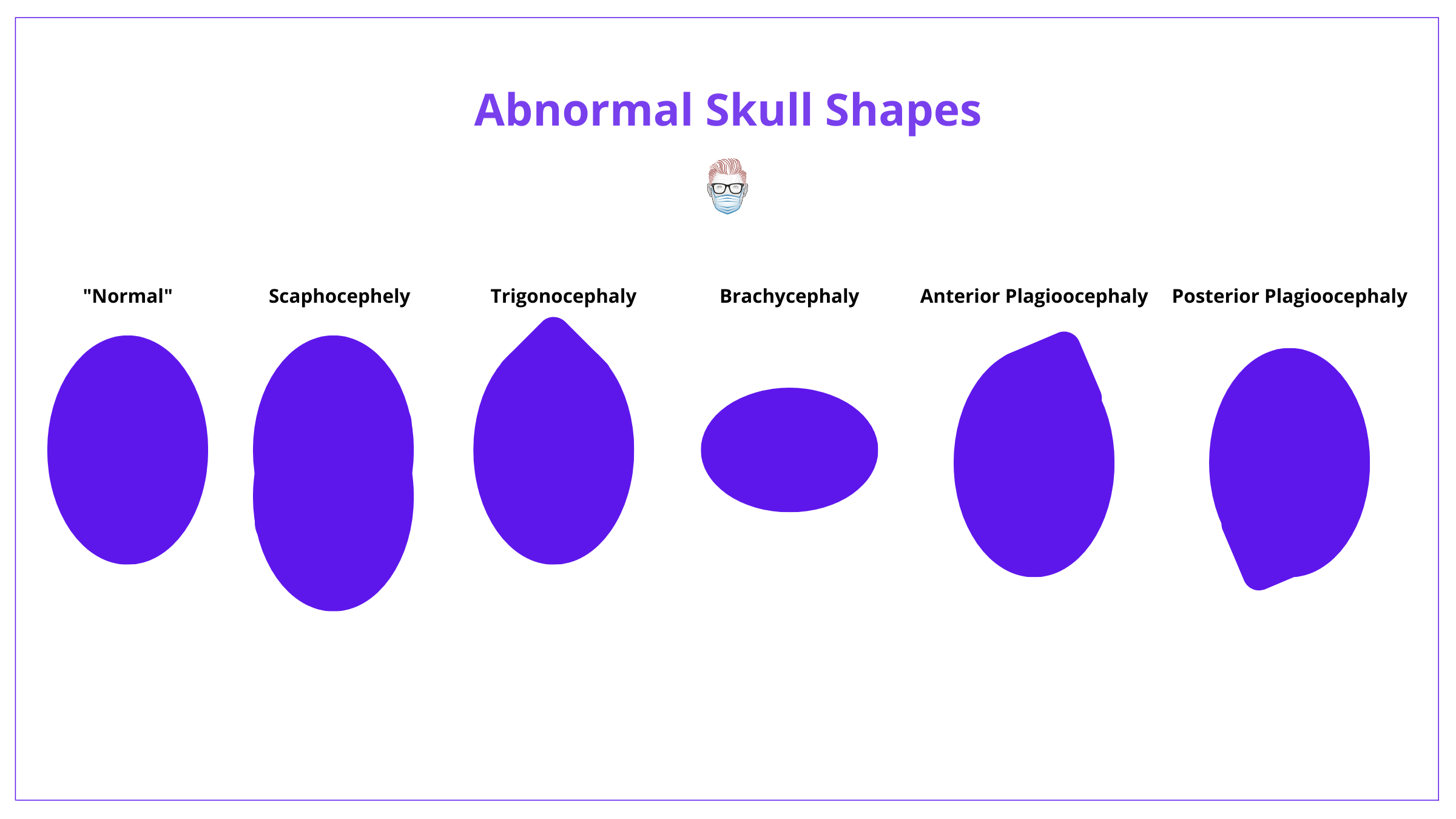
Craniosynostosis can be classified based on sutural involvement or head shape. These descriptions are correlated, as per Virchow Law.
Craniosynostosis can be classified by the affected suture(s) or head shape. This is because each pattern of fusion has a characteristic skull shape, as per Virchow's Law.
This classification is illustrated in the image detail below.
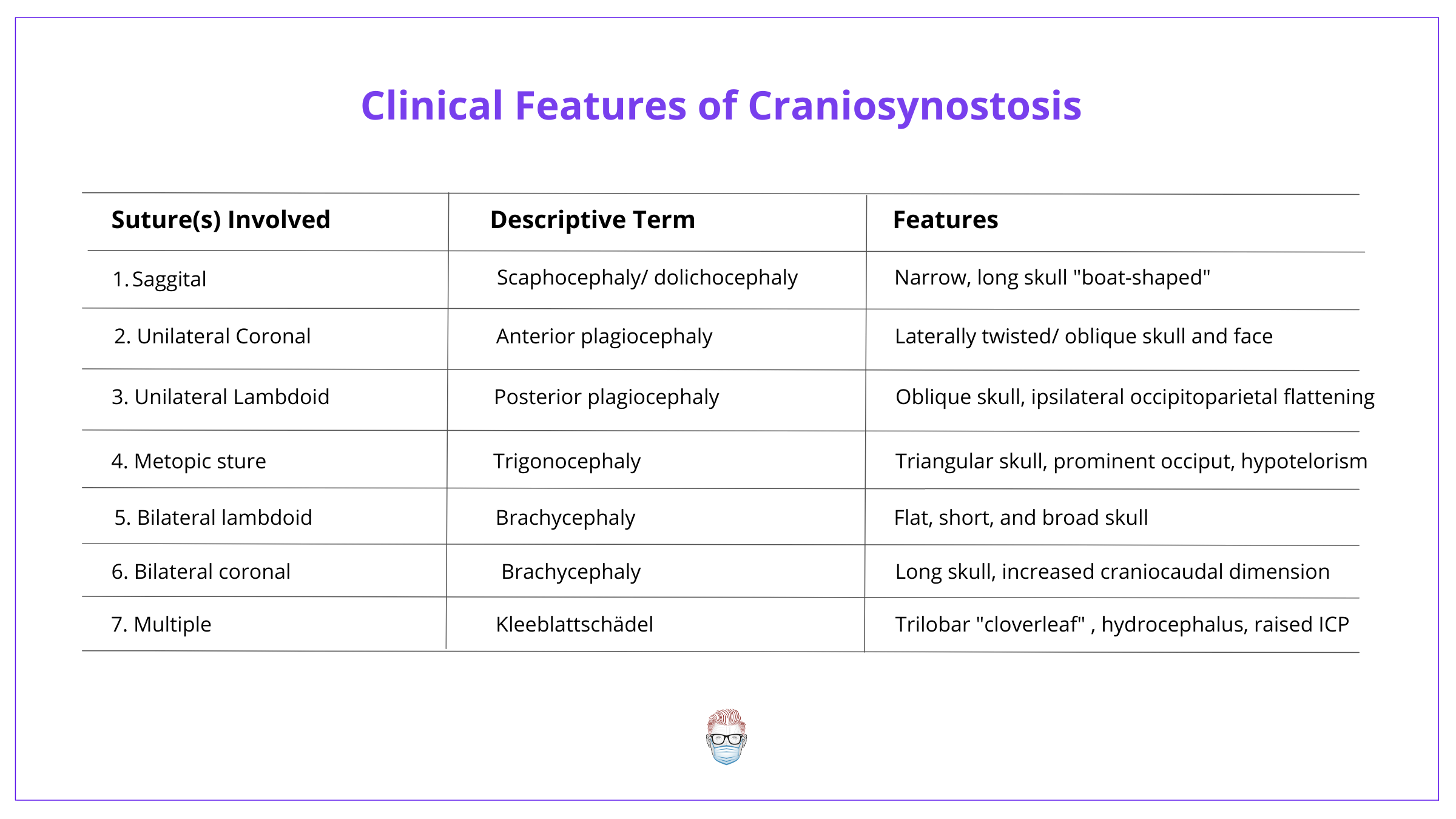
It can be difficult to remember the shapes of each condition. The best way is to understand Virchow's Law and use the following image.
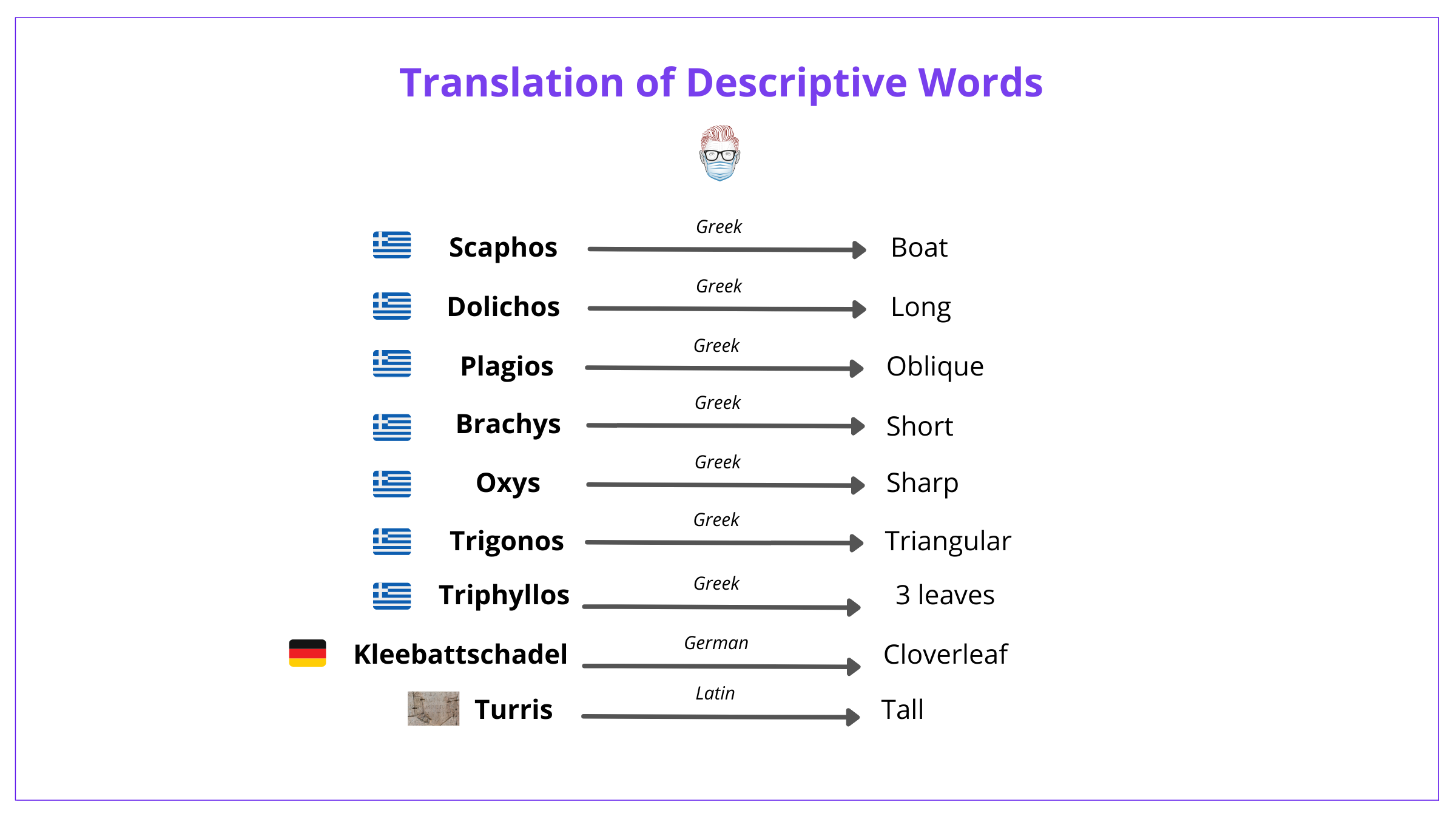
Another way is also to understand the translations of the words. These are detailed below.
Secondary Features of Craniosynostosis
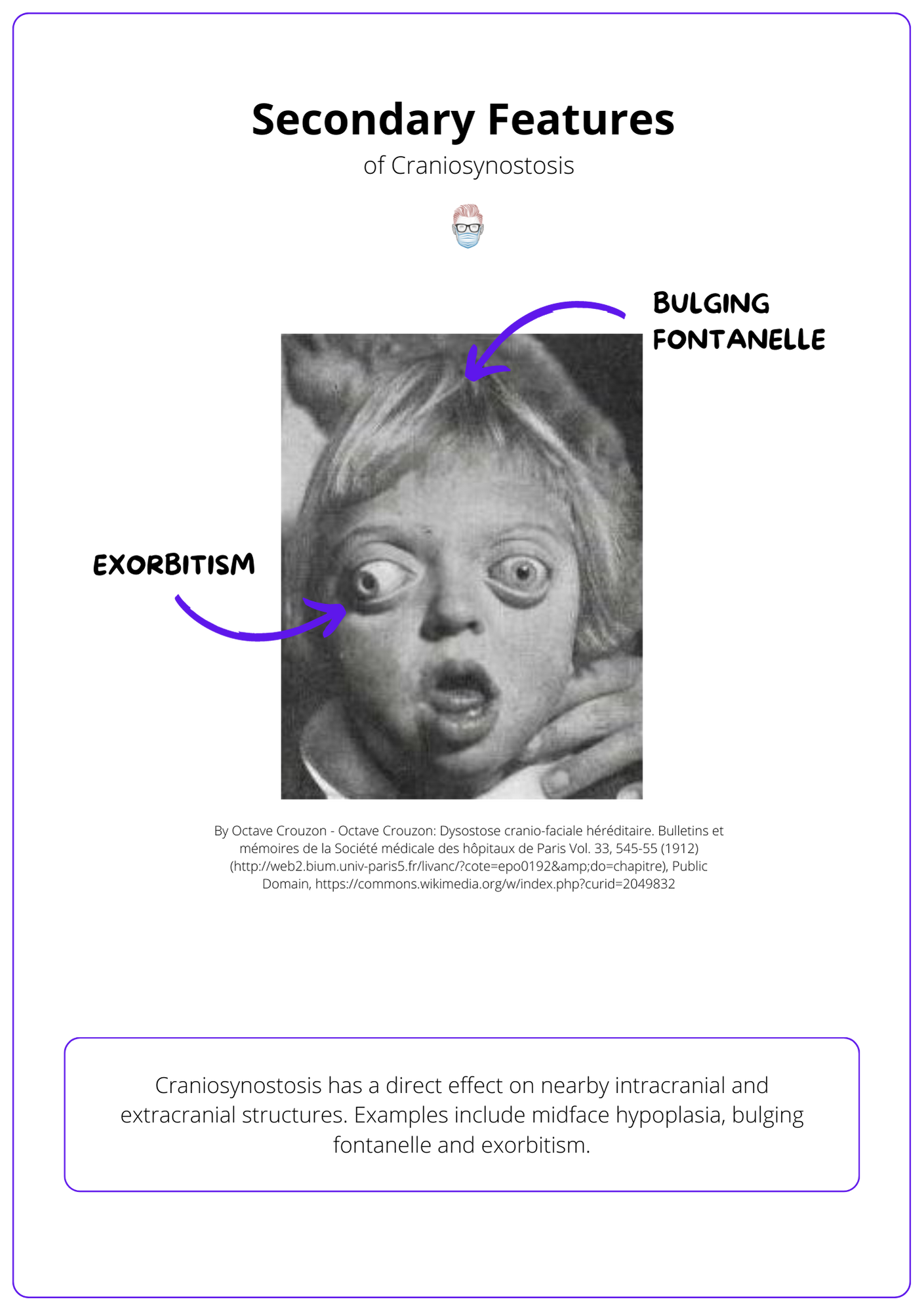
The premature fusion of sutures results in facial dysmorphology, associated limb abnormalities, and potentially raised intracranial pressure.
Craniosynostosis has a direct effect on nearby intracranial and extracranial structures.
- Midface Hypoplasia causing sleep apnea.
- Front-orbital rim recession causing exorbitism and exposure keratitis.
- Flare in the supraorbital rim found in trigonocephaly.
- Palpable ridges from the thickness of closed sutures. A metopic ridge can occur in trigonocephaly - it is benign and often self-resolving.
- Limb abnormalities are caused by a common molecular pathway.
- Raised intracranial pressure, which is discussed in more detail below.
Raised Intracranial Pressure
Craniosynostosis can result in raised intracranial pressures >15mmHg. This can often be linked to aqueductal stenosis.
These patients can have clinical features of:
- Head: headaches, irritability, seizures, bulging fontanelles (Volcano sign)
- Eyes: papilloedema, optic atrophy
- Abdomen: Vomiting
- Other: Developmental Delay
There is an increased risk when multiple suture synostosis. It can be the result of:
- Cephalocranial disproportion: decreased intracranial volume and restricted brain growth
- Vascular: Chiari Malformation, intracranial venous congestion
- Other: Hydrocephalus, upper Airway obstruction
Treatment considerations include a shunt, ventriculostomy, and craniovault remodeling.
Syndromic craniosynostosis
The majority of syndromic synostoses are autosomal dominant, caused by FGFR mutations, and have abnormal faces and limb abnormalities.
Overview
The majority are non-syndromic or isolated craniosynostosis. Only ~20% have genetic aetiology, which includes mutations in:
- Most common: FGFR mutations
- Less common: TWIST, TCF12, ERF, EFNB1, RAB23, POR, IL11RA.
This is described in the table below.

Features
Generally speaking, the syndromic conditions have similar features, which include:
- Autosomal dominant (except Carpenter and Antley-Bixter)
- Abnormal Facies
- Limb Abnormalities
Crouzon Syndrome (Most Common)
- Genetics: Autosomal dominant FGFR2 mutation (can occur sporadically).
- Synostosis: Bicoronal (other sutures can be involved).
- Features: Exorbitism, midface hypoplasia, high arched or cleft palate, conductive hearing loss, class III malocclusion.
- Sequelae: Progressive hydrocephalus.
Apert Syndrome
- Genetics: Autosomal dominant (although most cases are sporadic)
- Synostosis: Bicoronal
- Features: Parrot-beak, midface hypoplasia, cleft palate, syndactyly, class III malocclusion
- Sequelae: Raised ICP, nonprogressive ventriculomegaly, conductive loss
Carpenter's Syndrome (Rare)
- Genetics: Autosomal recessive RAB23
- Synostosis: usually sagittal or coronal, but many can be affected
- Features: cardiac issues, partial finger syndactyl, preaxial feet polydactyly
- Sequelae: obesity and intelligence
Pfeiffer Syndrome (Rare)
- Genetics: Autosomal dominant FGFR2 (most common & severe) or FGFR1.
- Clinical features: obstructive sleep apnea, broad thumbs/toes.
- Sequelae: can lead to Kleeblattschadel deformity.
Saethre-Chotzen Syndrome:
- Genetics: Autosomal dominant mutation in TWIST-1 gene
- Synostosis: Bicoronal
- Clinical features: low hairline, ptosis, prominent crus helicis, partial syndactyly, midface hypoplasia is less common
Muenke Syndrome:
- Genetics: Autosomal dominant FGFR3 mutation with variable expression
- Synostosis: coronal
- Clinical Features: developmental delay and sensorineural hearing loss
- NB: not all patients with Muenke syndrome suffer from craniosynostosis
Positional Plagiocephaly
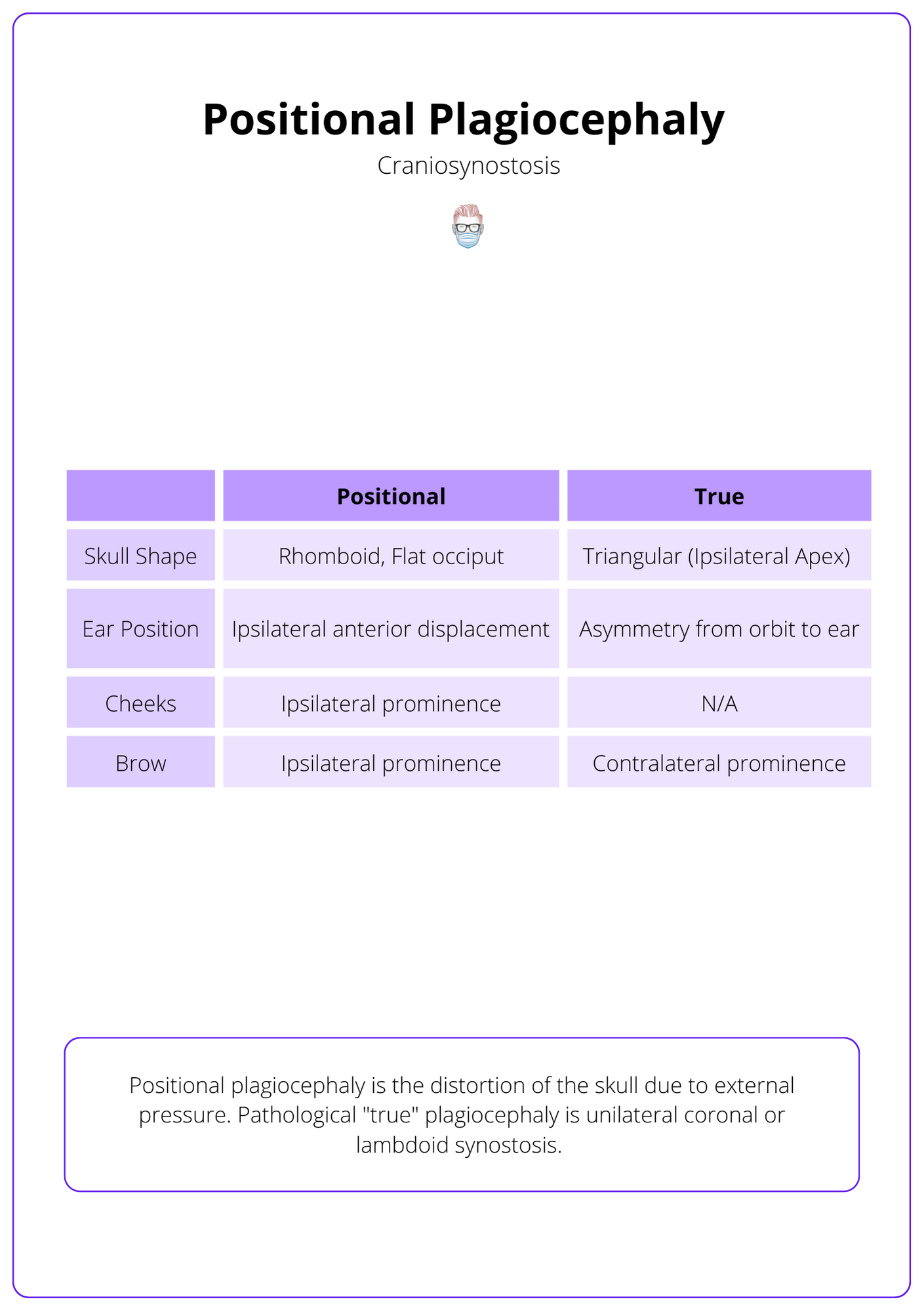
Positional plagiocephaly is external pressure resulting in a rhomboid-shaped head, and flat occiput with ipsilateral prominence of cheeks and brows.
Positional plagiocephaly is the distortion of the skull due to external pressure. Pathological "true" plagiocephaly is unilateral coronal or lambdoid synostosis.
These differences are illustrated in the table below.
Investigations
Radiological investigations such as X-ray, CT, or MRI can help confirm the clinical diagnosis, identify sutures, assess for complications, and plan for surgery.
Craniosynostosis is primarily a clinical diagnosis based on abnormal head shapes during the first year of life and secondary features. Investigations are used for diagnosis, pre-operative planning, and monitoring.
Radiology
There are different radiological investigations at the clinician's disposal.
Skull X-ray
This is an AP, lateral, Townes projection, and C-spine. Features include:
- Abnormal skull shape
- Suture sclerosis, loss of lucency, "lipping", or interdigitations
- Normal sutures widen to compensate
- Harlequin sign on the lateral orbit (coronal synostoses superiorly displacing the lesser wing of sphenoid)
- Copper-beaten skull from raised ICP
- "Thumb printing" from gyri pressure on the inner table
Cranial CT with 3D Reconstruction
- Assess each individual suture fusion, intracranial volume, and morphology.
- Useful for planning for surgery.
- Can assess for the presence of hydrocephalus.
- Raised ICP will result in reduced ventricle size and paracortical spaces, herniation through the foramen magnum.
The below image shows the radiological features of Craniosynostosis.
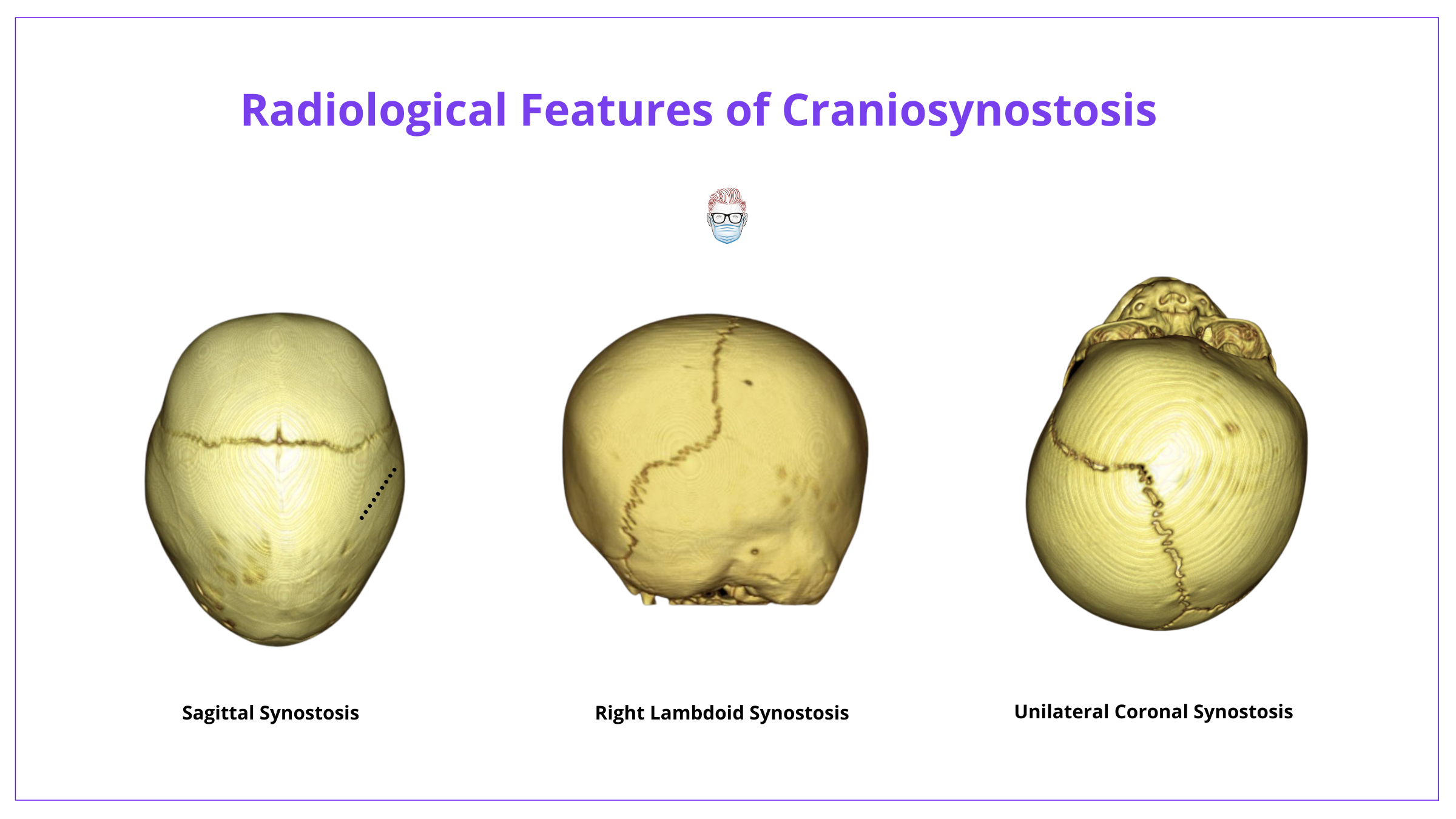
Transorbital Ultrasound
- Measure the diameter of the optic nerve sheath.
- Increased ICP in cases such as craniosynostosis usually leads to the increased diameter of the optic nerve sheath.
MRI
- Not routinely used.
- Can be considered for specific syndromes or concerns for specific pathologies, such as anomalous venous drainage.
Additional Investigations
Adjunctive investigations can help in certain circumstances. These include:
- Fundoscopy: assess for papilledema
- Direct Intraparenchymal monitoring: ICP monitoring
Managment of Craniosynostosis
Craniosyonotsis management focuses on the correction of shapes, treating complications, and maintaining functional status.
Overview
The treatment of craniosynostosis is centred on a multidisciplinary team involving a plastic surgeon, ENT, orthodontist, speech therapist, child psychologist, and nurses.
The goals of treatment should be to:
- To correct abnormal head shape/skull deformity.
- Treat or prevent raised intracranial pressure.
- Expand the intracranial volume.
- Protect the airway and cornea.
Surgical Options
A myriad of surgical options is available to treat craniosynostosis. The is no general consensus on the "best" time to perform surgery but generally is recommended in infancy between the ages of 3-12 months.
Options to consider include:
- Vault Remodelling and Reconstruction via distraction osteogenesis. This expansion technique can be performed at 6 months for Aperts, Crouzon's, Saethre-Chozen, and Meunke. TCVR can be performed via the 'Melbourne Technique' for sagittal synostosis via a wavy incision.
- Strip Craniectomy: strip excision of bone over suture. This can be considered in metopic synostosis or early sagittal synostosis.
- Fronto-orbital advancement increases AP dimension & reduces exorbitism. This can be performed in unicoronal synostosis.
- Monobloc advancement of Le Fort III and fronto-orbital section. This can be performed in extreme cases of raised ICP, exposure kerattis, and airway problems.
- Le Fort III to correct midface retrusion and secondary airway issues to become independent of tracheostomy and reduce sleep apnoea.
- Le Fort I for midface hypoplasia, class III malocclusion, and anterior open bite.
- Spring-assisted cranioplasty can be performed after a sagittal strip. It is a lower-risk and technically easier procedure but it doesn't fix frontal bossing.
- Skeletal distraction via internal or external devices ~1mm/day and usually for ~ 2-3 weeks. Regrowth primarily from the dura.
If hydrocephalus is present, shunting procedures are generally performed first, prior to bone remodeling.
Conclusion
1. Understanding of Craniosynostosis: You've gained a comprehensive understanding of the definition, causes, and Virchow's Law related to craniosynostosis.
2. Suture Anatomy: You've learned about the typical timeline and significance of the fusion of different cranial sutures, crucial for diagnosing and understanding the progression of craniosynostosis.
3. Classification Systems: You are now familiar with the different classifications of craniosynostosis based on affected sutures and resulting head shapes for the clinical assessment.
4. Secondary Features and Syndromic Variations: You've explored the secondary features associated with craniosynostosis, such as facial dysmorphology and limb abnormalities, as well as the distinctions between syndromic and nonsyndromic forms.
5. Diagnostic and Management Strategies: You've learned about the various diagnostic tools such as X-ray, CT, and MRI that confirm the diagnosis and assist in surgical planning, alongside a multidisciplinary approach to management, correcting deformities, and preventing complications.
Further Reading
- Kajdic N, Spazzapan P, Velnar T. Craniosynostosis - Recognition, clinical characteristics, and treatment. Bosn J Basic Med Sci. 2018 May 20;18(2):110-116. doi: 10.17305/bjbms.2017.2083. PMID: 28623672; PMCID: PMC5988529.
- Brah TK, Thind R, Abel DE. Craniosynostosis: Clinical Presentation, Genetics, and Prenatal Diagnosis. Obstet Gynecol Surv. 2020 Oct;75(10):636-644. doi: 10.1097/OGX.0000000000000830. PMID: 33111964.
- Kirmi O, Lo SJ, Johnson D, Anslow P. Craniosynostosis: a radiological and surgical perspective. Semin Ultrasound CT MR. 2009 Dec;30(6):492-512. doi: 10.1053/j.sult.2009.08.002. PMID: 20099636.
- Dempsey RF, Monson LA, Maricevich RS, Truong TA, Olarunnipa S, Lam SK, Dauser RC, Hollier LH Jr, Buchanan EP. Nonsyndromic Craniosynostosis. Clin Plast Surg. 2019 Apr;46(2):123-139. doi: 10.1016/j.cps.2018.11.001. Epub 2019 Jan 30. PMID: 30851746.
- Sawh-Martinez R, Steinbacher DM. Syndromic Craniosynostosis. Clin Plast Surg. 2019 Apr;46(2):141-155. doi: 10.1016/j.cps.2018.11.009. PMID: 30851747.
